---
title: History of how PMO Format was derived
---
<script>
$(document).ready(function() {
document.querySelectorAll('.downloadLink').forEach(function(e) { e.setAttribute('download', e.text); });
document.querySelectorAll('.downloadLink').forEach(function(e) { e.innerHTML = '<i class="fa fa-download"></i> ' + e.text; });
});
</script>
```{r setup, echo=FALSE}
source("../common.R")
```
# Sample and sequencing meta data fields
The meta fields for the sample and sequencing are based on NCBI naming schemes especially from SRA submission portal for human-associated pathogen. Depending on which reporting standard is selected at the time of submission, these are derived and vlided via the the [MiXS standards](https://genomicsstandardsconsortium.github.io/mixs/) set by the [Genomics Standards Consortium (GSC)](https://www.gensc.org/)
## SRA Sample fields
When submitting sample meta data to SRA, you have to choose a reporting standard to validate your meta data against
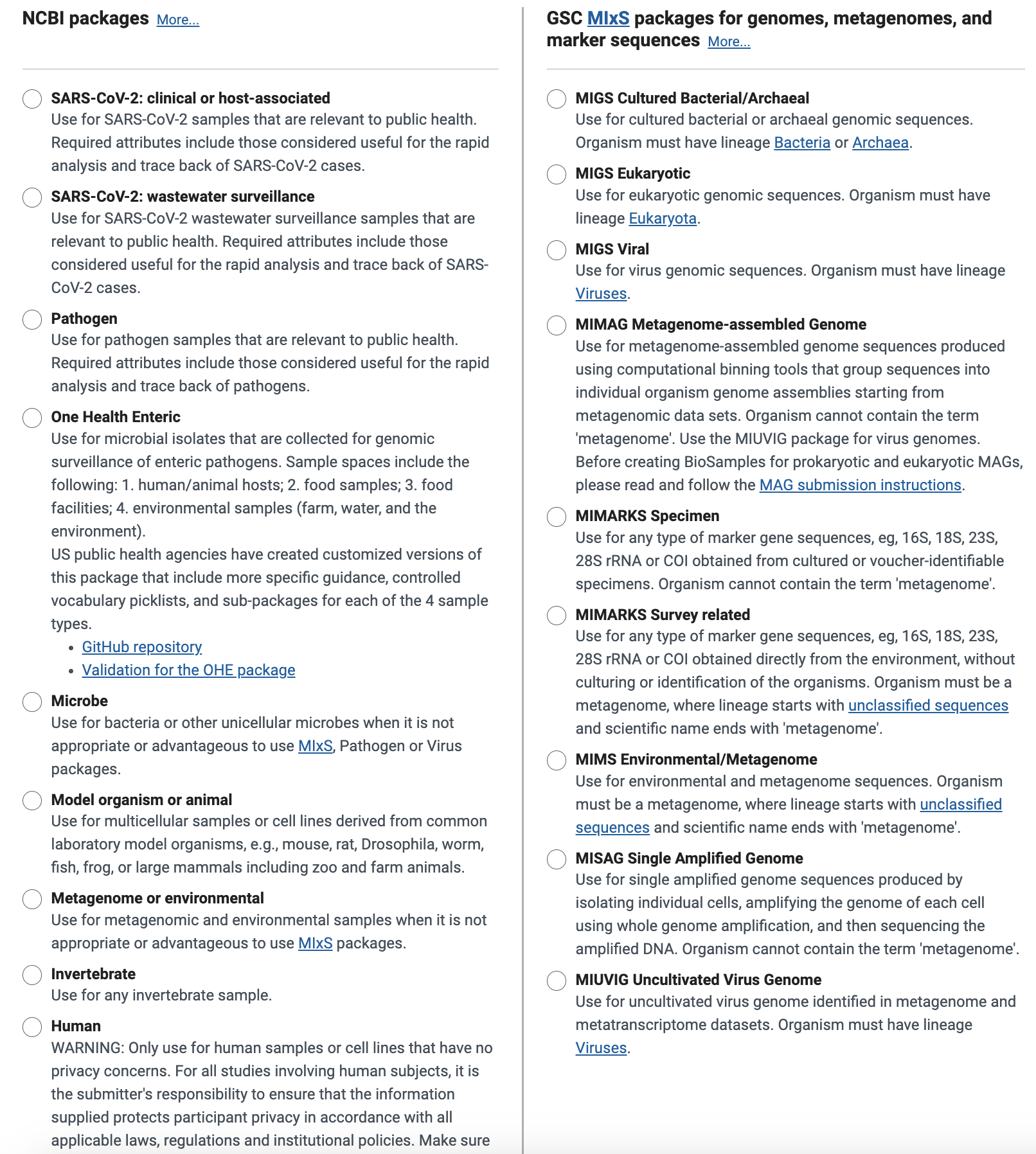
There are 4 reporting standards that one might pick for targeted amplicon sequencing of a pathogen
* [**MIGS.eu.human-associated.6.0**](#migs.eu.human-associated.6.0) (MiXS dervived/standard compliant, SRA encourages usage)
* [**MIMARKS.specimen.human-associated.6.0**](#mimarks.specimen.human-associated.6.0) (MiXS dervived/standard compliant, SRA encourages usage)
* [**Microbe.1.0**](#microbe.1.0) (non-MiXS derived, SRA discourages usage)
* [**Pathogen.cl.1.0**](#pathogen.cl.1.0) (non-MiXS derived, SRA discourages usage)
MIGS.eu.human-associated.6.0 and MIMARKS.specimen.human-associated.6.0 have the same required fields and only differ with a handful of different fields
Determining difference between MIGS.eu.human-associated and MIMARKS.specimen.human-associated
```{r}
MIGS_eu_human_associated = readxl::read_excel("sra_standards/MIGS.eu.human-associated.6.0.xlsx", skip = 12)
MIMARKS_specimen_human_associated = readxl::read_excel("sra_standards/MIMARKS.specimen.human-associated.6.0.xlsx", skip= 12)
colname_compared = set_decompose(colnames(MIGS_eu_human_associated), colnames(MIMARKS_specimen_human_associated))
list("Only in MIGS_eu_human_associated" = colname_compared$only_in_vectorA,
"Only in MIMARKS_specimen_human_associated" = colname_compared$only_in_vectorB)
```
Site describing all BioSample Attributes at the SRA <https://www.ncbi.nlm.nih.gov/biosample/docs/attributes/>
### MIGS.eu.human-associated.6.0
SRA website defining the fields: <https://submit.ncbi.nlm.nih.gov/biosample/template/?package-0=MIGS.eu.human-associated.6.0&action=definition>
GSC website defining the fields: <https://genomicsstandardsconsortium.github.io/mixs/0010002_0016003/>
Example file for SRA submission, these are all posible fields with required fields marked with a star(*)
```{r}
#| results: asis
#| echo: false
cat(createDownloadLink("sra_standards/MIGS.eu.human-associated.6.0.xlsx"))
```
```{r}
MIGS_eu_human_associated = readxl::read_excel("sra_standards/MIGS.eu.human-associated.6.0.xlsx", skip = 12)
create_dt(MIGS_eu_human_associated)
```
#### Required fields
Though all of these fields are required, they can also have the values of `not applicable`
* ***sample_name*** - Sample Name is a name that you choose for the sample. It can have any format, but we suggest that you make it concise, unique and consistent within your lab, and as informative as possible. Every Sample Name from a single Submitter must be unique
* ***organism*** - The most descriptive organism name for this sample (to the species, if possible). It is OK to submit an organism name that is not in our database. In the case of a new species, provide the desired organism name, and our taxonomists may assign a provisional taxID
* ***host*** (**non-MixS standard, equivalent is specific_host**)
* ***collection_date*** - the date on which the sample was collected; date/time ranges are supported by providing two dates from among the supported value formats, delimited by a forward-slash character; collection times are supported by adding "T", then the hour and minute after the date, and must be in Coordinated Universal Time (UTC), otherwise known as "Zulu Time" (Z); supported formats include "DD-Mmm-YYYY", "Mmm-YYYY", "YYYY" or ISO 8601 standard "YYYY-mm-dd", "YYYY-mm", "YYYY-mm-ddThh:mm:ss"; e.g., 30-Oct-1990, Oct-1990, 1990, 1990-10-30, 1990-10, 21-Oct-1952/15-Feb-1953, 2015-10-11T17:53:03Z; valid non-ISO dates will be automatically transformed to ISO format
* ***geo_loc_name*** - Geographical origin of the sample; use the appropriate name from this list https://www.insdc.org/submitting-standards/geo_loc_name-qualifier-vocabulary/. Use a colon to separate the country or ocean from more detailed information about the location, eg "Canada: Vancouver" or "Germany: halfway down Zugspitze, Alps"
* ***lat_lon*** - The geographical coordinates of the location where the sample was collected. Specify as degrees latitude and longitude in format "d[d.dddd] N|S d[dd.dddd] W|E", eg, 38.98 N 77.11 W
* ***env_broad_scale*** - Add terms that identify the major environment type(s) where your sample was collected. Recommend subclasses of biome [ENVO:00000428]. Multiple terms can be separated by one or more pipes e.g.: mangrove biome [ENVO:01000181]|estuarine biome [ENVO:01000020]
* ***env_local_scale*** - Add terms that identify environmental entities having causal influences upon the entity at time of sampling, multiple terms can be separated by pipes, e.g.: shoreline [ENVO:00000486]|intertidal zone [ENVO:00000316]
* ***env_medium*** - Add terms that identify the material displaced by the entity at time of sampling. Recommend subclasses of environmental material [ENVO:00010483]. Multiple terms can be separated by pipes e.g.: estuarine water [ENVO:01000301]|estuarine mud [ENVO:00002160]
* ***isol_growth_condt*** - PMID or url for isolation and growth condition specifications
One of the following fields, (none of these are technically a MixS standard but a MixS stand field called `subspecf_gen_lin` allows for the definiton of any of these)
* ***strain*** - microbial or eukaryotic strain name
* ***isolate*** - identification or description of the specific individual from which this sample was obtained
* ***cultivar*** - cultivar name - cultivated variety of plant
* ***ecotype*** - a population within a given species displaying genetically based, phenotypic traits that reflect adaptation to a local habitat, e.g., Columbia
### MIMARKS.specimen.human-associated.6.0
SRA website defining the fields: <https://submit.ncbi.nlm.nih.gov/biosample/template/?organism-organism_name=&organism-taxonomy_id=&package-0=MIMARKS.specimen&package-1=MIMARKS.specimen.human-associated.6.0&action=definition>
GSC website defining the fields: <https://genomicsstandardsconsortium.github.io/mixs/0010009_0016003/>
Example file for SRA submission, these are all posible fields with required fields marked with a star(*)
```{r}
#| results: asis
#| echo: false
cat(createDownloadLink("sra_standards/MIMARKS.specimen.human-associated.6.0.xlsx"))
```
```{r}
MIGS_eu_human_associated = readxl::read_excel("sra_standards/MIMARKS.specimen.human-associated.6.0.xlsx", skip = 12)
create_dt(MIGS_eu_human_associated)
```
#### Required fields
Though all of these fields are required, they can also have the values of `not applicable`
* ***sample_name*** - Sample Name is a name that you choose for the sample. It can have any format, but we suggest that you make it concise, unique and consistent within your lab, and as informative as possible. Every Sample Name from a single Submitter must be unique
* ***organism*** - The most descriptive organism name for this sample (to the species, if possible). It is OK to submit an organism name that is not in our database. In the case of a new species, provide the desired organism name, and our taxonomists may assign a provisional taxID
* ***host*** (**non-MixS standard, equivalent is specific_host**)
* ***collection_date*** - the date on which the sample was collected; date/time ranges are supported by providing two dates from among the supported value formats, delimited by a forward-slash character; collection times are supported by adding "T", then the hour and minute after the date, and must be in Coordinated Universal Time (UTC), otherwise known as "Zulu Time" (Z); supported formats include "DD-Mmm-YYYY", "Mmm-YYYY", "YYYY" or ISO 8601 standard "YYYY-mm-dd", "YYYY-mm", "YYYY-mm-ddThh:mm:ss"; e.g., 30-Oct-1990, Oct-1990, 1990, 1990-10-30, 1990-10, 21-Oct-1952/15-Feb-1953, 2015-10-11T17:53:03Z; valid non-ISO dates will be automatically transformed to ISO format
* ***geo_loc_name*** - Geographical origin of the sample; use the appropriate name from this list https://www.insdc.org/submitting-standards/geo_loc_name-qualifier-vocabulary/. Use a colon to separate the country or ocean from more detailed information about the location, eg "Canada: Vancouver" or "Germany: halfway down Zugspitze, Alps"
* ***lat_lon*** - The geographical coordinates of the location where the sample was collected. Specify as degrees latitude and longitude in format "d[d.dddd] N|S d[dd.dddd] W|E", eg, 38.98 N 77.11 W
* ***env_broad_scale*** - Add terms that identify the major environment type(s) where your sample was collected. Recommend subclasses of biome [ENVO:00000428]. Multiple terms can be separated by one or more pipes e.g.: mangrove biome [ENVO:01000181]|estuarine biome [ENVO:01000020]
* ***env_local_scale*** - Add terms that identify environmental entities having causal influences upon the entity at time of sampling, multiple terms can be separated by pipes, e.g.: shoreline [ENVO:00000486]|intertidal zone [ENVO:00000316]
* ***env_medium*** - Add terms that identify the material displaced by the entity at time of sampling. Recommend subclasses of environmental material [ENVO:00010483]. Multiple terms can be separated by pipes e.g.: estuarine water [ENVO:01000301]|estuarine mud [ENVO:00002160]
* ***isol_growth_condt*** - PMID or url for isolation and growth condition specifications
One of the following fields, (none of these are technically a MixS standard but a MixS stand field called `subspecf_gen_lin` allows for the definiton of any of these)
* ***strain*** - microbial or eukaryotic strain name
* ***isolate*** - identification or description of the specific individual from which this sample was obtained
* ***cultivar*** - cultivar name - cultivated variety of plant
* ***ecotype*** - a population within a given species displaying genetically based, phenotypic traits that reflect adaptation to a local habitat, e.g., Columbia
### Microbe.1.0
SRA website defining the fields: <https://submit.ncbi.nlm.nih.gov/biosample/template/?organism-organism_name=&organism-taxonomy_id=&package-0=Microbe.1.0&action=definition>
Example file for SRA submission, these are all posible fields with required fields marked with a star(*)
```{r}
#| results: asis
#| echo: false
cat(createDownloadLink("sra_standards/Microbe.1.0.xlsx"))
```
```{r}
Microbe = readxl::read_excel("sra_standards/Microbe.1.0.xlsx", skip = 12)
create_dt(Microbe)
```
#### Required fields
Though all of these fields are required, they can also have the values of `not applicable`
* ***sample_name*** - Sample Name is a name that you choose for the sample. It can have any format, but we suggest that you make it concise, unique and consistent within your lab, and as informative as possible. Every Sample Name from a single Submitter must be unique
* ***organism*** - The most descriptive organism name for this sample (to the species, if possible). It is OK to submit an organism name that is not in our database. In the case of a new species, provide the desired organism name, and our taxonomists may assign a provisional taxID
* ***collection_date*** - the date on which the sample was collected; date/time ranges are supported by providing two dates from among the supported value formats, delimited by a forward-slash character; collection times are supported by adding "T", then the hour and minute after the date, and must be in Coordinated Universal Time (UTC), otherwise known as "Zulu Time" (Z); supported formats include "DD-Mmm-YYYY", "Mmm-YYYY", "YYYY" or ISO 8601 standard "YYYY-mm-dd", "YYYY-mm", "YYYY-mm-ddThh:mm:ss"; e.g., 30-Oct-1990, Oct-1990, 1990, 1990-10-30, 1990-10, 21-Oct-1952/15-Feb-1953, 2015-10-11T17:53:03Z; valid non-ISO dates will be automatically transformed to ISO format
* ***geo_loc_name*** - Geographical origin of the sample; use the appropriate name from this list https://www.insdc.org/submitting-standards/geo_loc_name-qualifier-vocabulary/. Use a colon to separate the country or ocean from more detailed information about the location, eg "Canada: Vancouver" or "Germany: halfway down Zugspitze, Alps"
* ***sample_type*** (**non MixS standard**) - Sample type, such as cell culture, mixed culture, tissue sample, whole organism, single cell, metagenomic assembly
One of the following (**none of these are technically a MixS standard but a MixS stand field called** `subspecf_gen_lin` **allows for the definiton of any of these**)
* ***strain*** - microbial or eukaryotic strain name
* ***isolate*** - identification or description of the specific individual from which this sample was obtained
One of the following
* ***host*** (**non-MixS standard, equivalent is specific_host**) - The natural (as opposed to laboratory) host to the organism from which the sample was obtained. Use the full taxonomic name, eg, "Homo sapiens".
* ***isolation_source*** (**non MixS standard**) - Describes the physical, environmental and/or local geographical source of the biological sample from which the sample was derived
### Pathogen.cl.1.0
SRA website defining the fields: <https://submit.ncbi.nlm.nih.gov/biosample/template/?organism-organism_name=&organism-taxonomy_id=&package-0=Pathogen&package-1=Pathogen.cl.1.0&action=definition>
Example file for SRA submission, these are all posible fields with required fields marked with a star(*)
```{r}
#| results: asis
#| echo: false
cat(createDownloadLink("sra_standards/Pathogen.cl.1.0.xlsx"))
```
```{r}
Pathogen = readxl::read_excel("sra_standards/Pathogen.cl.1.0.xlsx", skip = 12)
create_dt(Pathogen)
```
#### Required fields
Though all of these fields are required, they can also have the values of `not applicable`
* ***sample_name*** - Sample Name is a name that you choose for the sample. It can have any format, but we suggest that you make it concise, unique and consistent within your lab, and as informative as possible. Every Sample Name from a single Submitter must be unique
* ***organism*** - The most descriptive organism name for this sample (to the species, if possible). It is OK to submit an organism name that is not in our database. In the case of a new species, provide the desired organism name, and our taxonomists may assign a provisional taxID
* ***collection_date*** - the date on which the sample was collected; date/time ranges are supported by providing two dates from among the supported value formats, delimited by a forward-slash character; collection times are supported by adding "T", then the hour and minute after the date, and must be in Coordinated Universal Time (UTC), otherwise known as "Zulu Time" (Z); supported formats include "DD-Mmm-YYYY", "Mmm-YYYY", "YYYY" or ISO 8601 standard "YYYY-mm-dd", "YYYY-mm", "YYYY-mm-ddThh:mm:ss"; e.g., 30-Oct-1990, Oct-1990, 1990, 1990-10-30, 1990-10, 21-Oct-1952/15-Feb-1953, 2015-10-11T17:53:03Z; valid non-ISO dates will be automatically transformed to ISO format
* ***geo_loc_name*** - Geographical origin of the sample; use the appropriate name from this list https://www.insdc.org/submitting-standards/geo_loc_name-qualifier-vocabulary/. Use a colon to separate the country or ocean from more detailed information about the location, eg "Canada: Vancouver" or "Germany: halfway down Zugspitze, Alps"
* ***collected_by*** - Name of persons or institute who collected the sample
* ***host*** (**non-MixS standard, equivalent is specific_host**) - The natural (as opposed to laboratory) host to the organism from which the sample was obtained. Use the full taxonomic name, eg, "Homo sapiens".
* ***host_disease*** (**non-MixS field**) - Name of relevant disease, e.g. Salmonella gastroenteritis. Controlled vocabulary, http://bioportal.bioontology.org/ontologies/1009 or http://www.ncbi.nlm.nih.gov/mesh
* ***isolation_source*** (**non MixS standard**) - Describes the physical, environmental and/or local geographical source of the biological sample from which the sample was derived
* ***lat_lon*** - The geographical coordinates of the location where the sample was collected. Specify as degrees latitude and longitude in format "d[d.dddd] N|S d[dd.dddd] W|E", eg, 38.98 N 77.11 W
One of the following (**none of these are technically a MixS standard but a MixS stand field called** `subspecf_gen_lin` **allows for the definiton of any of these**)
* ***strain*** - microbial or eukaryotic strain name
* ***isolate*** - identification or description of the specific individual from which this sample was obtained
### Additional details
Additonal fields not in GSC or SRA submission but are often found in downloads from SRAs. This is because the geo_loc_name can be country followed by several more details, these columns allow for listing only the country and continent
* ***geo_loc_name_country***
* ***geo_loc_name_country_continent***
## SRA Sequencing fields
Info about the sequencing of the samples above, details can be found below link:
SRA sequencing meta: <https://www.ncbi.nlm.nih.gov/sra/docs/submitportal/#6-sra-metadata>
Example file for SRA submission
```{r}
#| results: asis
#| echo: false
cat(createDownloadLink("sra_standards/SRA_metadata.xlsx"))
```
```{r}
SRA_sequencing_metadata = readxl::read_excel("sra_standards/SRA_metadata.xlsx", sheet = 2)
create_dt(SRA_sequencing_metadata)
```
### Required fields
Details about the requirments
* If you created samples previously, provide accessions in the form of SAMN# in the column sample_accession. Otherwise provide the sample name used in the BioSample attributes spreadsheet.
* Each row in the template represents a sequencing library with a unique combination of sample + library + sequencing strategy + layout + instrument model. Each row should have a unique library_id that is short and meaningful (like an ID you might use in lab).
* When libraries are indeed identical (same combination of sample + library + strategy + layout + instrument model), all files should be placed in the same row To do this simply enter the file names consecutively in the same row by adding more columns with headers filename2, filename3, etc…. PAIRED files must always be listed in the same row.
* ***sample_name*** - must match exactly the **sample_name** in the tables above
* ***library_id*** - each must be unique, should be short like what is in a samplesheet
* ***title*** - Short description that will identify the dataset on public pages. A clear and concise formula for the title would be like: {methodology} of {organism}: {sample info} _e.g. RNA-Seq of mus musculus:adult female spleen
* ***library_strategy*** - what the nuceloacid sequencing/amplification strategy was (common names are AMPLICON, WGS)
* ***library_source*** - Source of amplification material (common names GENOMIC, TRANSCRIPTOMIC)
* ***library_selection*** - how amplification was done (common are PCR=Source material was selected by designed primers, RANDOM =Random selection by shearing or other method)
* ***library_layout*** (**MixS equivalent lib_layout**) - Specify whether to expect single, paired, or other configuration of reads
* ***platform*** (**MixS equivalent is part of seq_meth**) - Machine used to sequence data, should be one from <https://ontobee.org/ontology/OBI?iri=http://purl.obolibrary.org/obo/OBI_0400103>
* ***instrument_model*** (**MixS equivalent is part of seq_meth**) - The specific model of the machine above
* ***design_description*** - A short description of how sequencing was done, paragraph style
* filetype and filenames - the type of file and the names of the files associated with the sequencing
```{r, eval = F, echo = F}
PRJNA1180199_SRR31271348_SraRunTable = readr::read_csv("sra_standards/examples_sra_download_info/PRJNA1180199_SRR31271348_SraRunTable.csv")
PRJNA1180199_SRR31271447_SraRunTable = readr::read_csv("sra_standards/examples_sra_download_info/PRJNA1180199_SRR31271447_SraRunTable.csv")
set_decompose(colnames(PRJNA1180199_SRR31271348_SraRunTable), colnames(PRJNA1180199_SRR31271447_SraRunTable))
PRJNA1180199_allRunInfo = readr::read_tsv("sra_standards/examples_sra_download_info/PRJNA1180199_allRunInfo.tsv")
```
## GSC MiXS standards
### MIGS.eu
<https://genomicsstandardsconsortium.github.io/mixs/0010002/>
```{r}
MigsEu_v6.2.0_fields = readr::read_tsv("mixs_standards/MigsEu_v6.2.0_fields.txt") %>%
arrange(fields)
create_dt(MigsEu_v6.2.0_fields)
MigsEu_v6.2.0_fields = MigsEu_v6.2.0_fields%>%
mutate(in_MigsEu_v6.2.0 = T)
```
### MIGS.human-associated
<https://genomicsstandardsconsortium.github.io/mixs/0016003/>
```{r}
HumanAssociated_v6.2.0_fields = readr::read_tsv("mixs_standards/HumanAssociated_v6.2.0_fields.txt") %>%
arrange(fields)
create_dt(HumanAssociated_v6.2.0_fields)
HumanAssociated_v6.2.0_fields = HumanAssociated_v6.2.0_fields%>%
mutate(in_HumanAssociated_v6.2.0 = T)
```
### MIGS.eu.human-associated
<https://genomicsstandardsconsortium.github.io/mixs/0010002_0016003/>
### MIMARKS.specimen
<https://genomicsstandardsconsortium.github.io/mixs/0010009/>
```{r}
MimarksC_v6.2.0_fields = readr::read_tsv("mixs_standards/MimarksC_v6.2.0_fields.txt") %>%
arrange(fields)
create_dt(MimarksC_v6.2.0_fields)
MimarksC_v6.2.0_fields = MimarksC_v6.2.0_fields%>%
mutate(in_MimarksC_v6.2.0 = T)
```
### MIMARKS.specimen.human-associated
<https://genomicsstandardsconsortium.github.io/mixs/0010009_0016003/>
### All fields
```{r}
all_mixs_fields = MigsEu_v6.2.0_fields %>%
full_join(MimarksC_v6.2.0_fields) %>%
full_join(HumanAssociated_v6.2.0_fields) %>%
arrange(fields) %>%
mutate(in_both_MigsEu_MimarksC = in_MigsEu_v6.2.0 & in_MimarksC_v6.2.0)%>%
mutate(in_all_three = in_MigsEu_v6.2.0 & in_MimarksC_v6.2.0 & in_HumanAssociated_v6.2.0)
create_dt(all_mixs_fields)
```
## PMO Sample and sequencing fields
The fields choices for PMO took into consideration the Mixs standards, SRA standards, and from other similar amplicon standards (which are also based on Mixs standards), [Environmental System Science Data Infrastructure for a Virtual Ecosystem (ESS-DIVE)](https://github.com/ess-dive-workspace/essdive-amplicon/tree/main) and [National Microbiome Data Collaborative (NMDC)](https://github.com/microbiomedata/nmdc-schema?tab=readme-ov-file)
The SRA takes two tables, one defining bio samples and one defining sequencing experiment libraries done on those bio samples of which there could be multiple sequencing experiments. Therefore the decision was made to also create two separate data sections for defining a biosample ([specimen_info](FormatOverviewAdvanced.qmd#specimeninfo)) and one defining experiments on those specimens ([library_sample_info](FormatOverviewAdvanced.qmd#librarysampleinfo)), this allows the mirroring of SRA as well as allowing for storing replicates of a single specimen. A 3rd section was created to store redundant sequencing info ([sequencing_info](FormatOverviewAdvanced.qmd#sequencinginfo)).
### specimen_info
```{r, echo =F, eval=F}
pmo_schema = rjson::fromJSON(file = "portable_microhaplotype_object.schema.json")
cat(sort(pmo_schema$`$defs`$SpecimenInfo$required), sep = "\n")
cat(sort(names(pmo_schema$`$defs`$SpecimenInfo$properties)[names(pmo_schema$`$defs`$SpecimenInfo$properties) %!in% pmo_schema$`$defs`$SpecimenInfo$required]), sep= "\n")
```
Comparing the specimen_info fields to the MIXS standard and SRA. Please see [specimen_info in FormatOverview](FormatOverviewAdvanced.qmd#specimeninfo) for short description of each pmo field and [above](#required-fields) for the SRA fields
```{r}
create_dt(readr::read_tsv("specimen_info_field_comparison.tsv"))
```
```{r, echo=FALSE, eval=FALSE}
specimen_info_field_comparison = readr::read_tsv("specimen_info_field_comparison.tsv")
specimen_info_field_comparison = specimen_info_field_comparison %>%
mutate(only_in_pmo = is.na(`MIXS v6.2.0 MigsEu/HumanAssociated/MimarksC`) & is.na(`SRA MigsEu/HumanAssociated/MimarksC`))
specimen_info_field_comparison %>% group_by(pmo_required, only_in_pmo) %>% count()%>%
mutate(total_pmo = specimen_info_field_comparison %>% filter(!is.na(pmo_required)) %>% nrow())
```
### sequencing_info and library_sample_info
```{r, echo =F, eval=F}
pmo_schema = rjson::fromJSON(file = "portable_microhaplotype_object.schema.json")
cat(sort(pmo_schema$`$defs`$LibrarySampleInfo$required), sep = "\n")
cat(sort(names(pmo_schema$`$defs`$LibrarySampleInfo$properties)[names(pmo_schema$`$defs`$LibrarySampleInfo$properties) %!in% pmo_schema$`$defs`$LibrarySampleInfo$required]), sep= "\n")
cat(sort(pmo_schema$`$defs`$SequencingInfo$required), sep = "\n")
cat(sort(names(pmo_schema$`$defs`$SequencingInfo$properties)[names(pmo_schema$`$defs`$SequencingInfo$properties) %!in% pmo_schema$`$defs`$SequencingInfo$required]), sep= "\n")
```
Comparing the [sequencing_info](FormatOverviewAdvanced.qmd#sequencinginfo) and [library_sample_info](FormatOverviewAdvanced.qmd#librarysampleinfo) fields to the Mixs standards and the data required for SRA submission [above](#required-fields-4)
```{r}
create_dt(readr::read_tsv("library_and_sequencing_field_comparison.tsv"))
```
```{r, echo=FALSE, eval=FALSE}
library_and_sequencing_field_comparison = readr::read_tsv("library_and_sequencing_field_comparison.tsv")
library_and_sequencing_field_comparison = library_and_sequencing_field_comparison %>%
mutate(only_in_pmo = is.na(`MIXS v6.2.0 MigsEu/HumanAssociated/MimarksC`) & is.na(`SRA MigsEu/HumanAssociated/MimarksC`))
library_and_sequencing_field_comparison %>% group_by(pmo_required, only_in_pmo) %>% count() %>%
mutate(total_pmo = library_and_sequencing_field_comparison %>% filter(!is.na(pmo_required)) %>% nrow())
```
{{< fa dna >}}
