Portable Microhaplotype Object (PMO)
Multiplexed targeted sequencing is now widely used to generate data for the most informative genomic regions of organisms, but the lack of an appropriate data standard has hindered data sharing, reuse, and downstream analysis. Here, we provide details for an extensible standard and related convenience utilities to store lossless, compact representations of phased, processed target sequences (microhaplotypes) along with an efficient relational ontology in a portable JSON file.
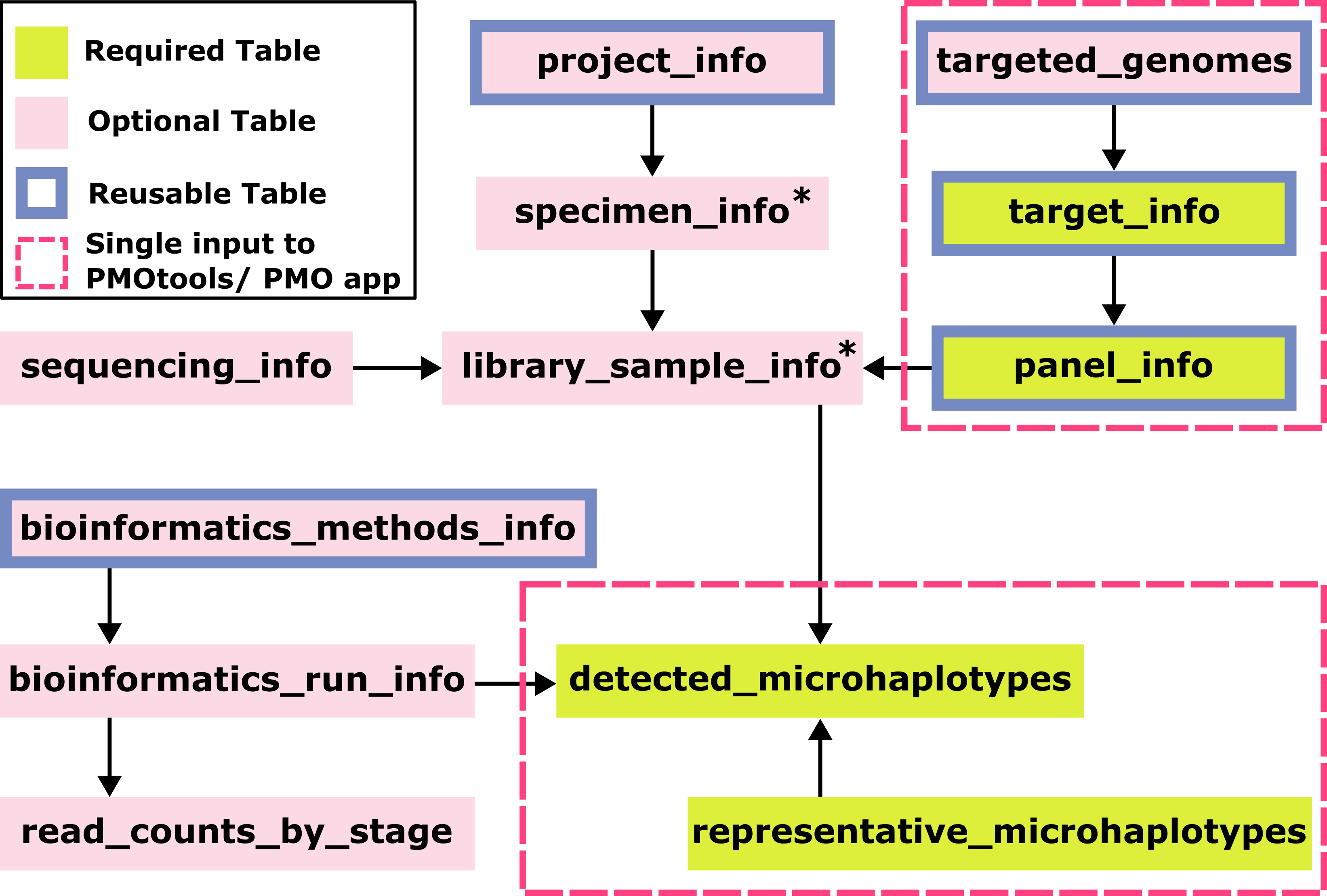 * specimen_info and library_sample_info only need identifiers which can be autogenerated from the detected_microhalotypes
* specimen_info and library_sample_info only need identifiers which can be autogenerated from the detected_microhalotypes
Quick Start
To get started, you can use the PMO App to build a PMO or use the pmotools-python package to build a PMO. We recommend using the PMO App if you don’t have any programming/ Python experience and the pmotools-python package for more advanced usage.
PMO App documentation pmotools-python documentation
If you are new to building a PMO, the template below can be a useful starting point.
Motivation
Targeted amplicon sequencing is now established as a sensitive and efficient means of obtaining relevant information about a wide variety of organisms. Applications are broad and expanding, including microbiome analysis, pathogen identification, detection of antimicrobial resistance, and tracking the spread of viruses, bacteria, and eukaryotic pathogens.
Many of these applications utilize the full sequences provided by individual reads because they contain multiple, phased variants (microhaplotypes)(Oldoni et al. 2019) - information that is lost when decomposing these data into independent variants such as SNPs. This information is particularly valuable when sequencing samples containing organisms with more than one sequence per target, such as mixed bacterial samples, commonly polyclonal pathogens (e.g., Plasmodium) (Tessema et al. 2022),(LaVerriere et al. 2022),(Jacob et al. 2021),(Kattenberg Johanna Helena et al. 2023),(Aranda-Díaz et al. 2025),(Sadler et al. 2024), and diploid or polyploid organisms. Thus, data formats designed for small variants that do not preserve full sequences, such as the popular variant call format (VCF), are not well suited to store microhaplotype data.
Objectives
- Provide a structured and flexible framework to help individual researchers and groups to organize their data in a findable and accessible way.
- Create a standard for data sharing, including for repositories, academic reports, and public health entities, to aid in interoperability, transparency, and reproducibility
- Maximize data reuse by lowering the barriers for making data publicly available in a standardized format.
- Provide a consistent format to allow harmonization of downstream analysis tools across data sets and minimize the need for tedious and error-prone tasks such as data reshaping
PMO structure and ontology
Please see this page for details on the internal structure of a PMO and its naming schema.
Convenience Tools
There are two ways to build a PMO using our tools:
- PMO App - An interactive, no-code PMO builder. The first load may take a moment. The app runs entirely in your browser, and all data remain on your computer—nothing is uploaded.
- pmotools-python - Build and work with PMO files in Python or from the command line. See the documentation and tutorials.
PMO in the Wider Ecosystem
PMO serves as a convergence point within the broader data ecosystem between sample collection/allele calling and various downstream analyses. Please see here for more details
Development Approach
The microbiome community has created data standards for a single locus, including BIOM and ESS-DIVE. Here, we extend these standards to an arbitrary number of loci in a framework extensible to any type of targeted sequence data. The format is lossless, allowing recovery of full sequence data, while achieving data compression of ~6x and up to ~80x with additional compression using standard tools (e.g., gzip). Optional fields allow data generators with domain expertise to include additionally processed sequence data such as variants with masked domains, e.g. for highly error prone areas such as tandem repeats. Notably, the framework provides a robust relational ontology for sample, laboratory, and bioinformatic metadata in addition to sequence data, mitigating the common problem of partially or completely orphaned data. A full ontology has been built out for Plasmodium, leveraging existing fields where available, and the modular structure can be flexibly extended to other biological systems, including those containing multiple types of organisms. All data are encoded in a standard JSON file, enhancing portability and ease-of-interpretation. The end result is a design which is efficient, lightweight, and flexible, organizing metadata together with genetic data. Finally, we have created a set of convenience utilities to make it easy to create, manipulate, share, import, and export PMO files.
Please see here for a detailed breakdown of how the fields chosen relate to other standards
Community Engagement
The development of the PMO format would not be possible without the community engagement and feedback that we have received and we want to continue to incorporate feedback while we maintain the format. Please reach out to info@plasmogenepi.org with any general questions or feedback.
If you have questions on the documentation of the format, you can use the github issues page to ask questions, post suggestions for both the documentation and format: https://github.com/PlasmoGenEpi/PMO_Docs/issues
If you have questions on the python implementation of interacting with the format, you can use github issues page here: https://github.com/PlasmoGenEpi/pmotools-python/issues